
 Fact-checked by
Fact-checked by  Discussion
Discussion
Friedreich’s ataxia causes and risk factors
Friedreich’s ataxia (FA), a rare genetic disease affecting the nerves and muscles, is caused by mutations in the FXN gene. The condition develops when a person carries two mutated copies of the gene, one inherited from each biological parent.
These mutations lead to a lack of production of frataxin, a protein important for the function of mitochondria, the cellular energy production centers.
Nerves and muscles are particularly vulnerable to the loss of cellular energy that accompanies frataxin deficiency, leading to the core symptoms of FA, which may include problems with coordination and balance (ataxia), muscle weakness, heart problems, and neurological issues.
What causes FA?
FA is caused by mutations in both copies of the FXN gene. This is the only cause of FA. In the overwhelming majority of cases — more than 95% — the type of mutation found in both gene copies is called a GAA trinucleotide repeat expansion.
Genes are made up of long strings of building blocks, or nucleotides, that code for a specific protein. There are four types of nucleotides: adenine (A), cytosine (C), guanine (G), and thymine (T).
The FXN gene contains a section of DNA wherein three nucleotides — GAA — are repeated several times, normally about five to 33 times. However, this sequence is repeated many more times in people with FA, ranging from 66 to more than 1,000.
Less frequently, people living with FA will have this type of mutation in one copy of the FXN gene, and another type of mutation, called a point mutation, in the other copy. There, only a single nucleotide is changed, deleted, or inserted abnormally.
Either way, FA-causing genetic alterations mean that FXN‘s ability to make frataxin is disrupted, leading to a severe reduction in the protein’s levels.
While frataxin’s exact biological roles are not entirely understood, it is generally accepted that it is required for proper functioning of mitochondria, cellular structures that produce most of the energy needed by cells to function.
Particularly, frataxin helps to regulate iron metabolism in cells, and is important for the production of certain iron-containing compounds that mitochondria use to function. Ultimately, the consequence of frataxin deficiency in FA is a disruption in cellular energy production.
Frataxin loss also is linked to a type of cellular damage called oxidative stress that drives cell death.
Nerve and muscle cells, which have high energy demands, are particularly susceptible to frataxin loss. Damage to these cell types is primarily what leads to the symptoms of Friedreich’s ataxia.
Because a higher number of GAA repeats is linked to greater frataxin deficiencies, patients who have longer repeats may have a younger age at disease onset and more severe or rapidly progressing symptoms relative to patients with fewer repeats.
FA inheritance
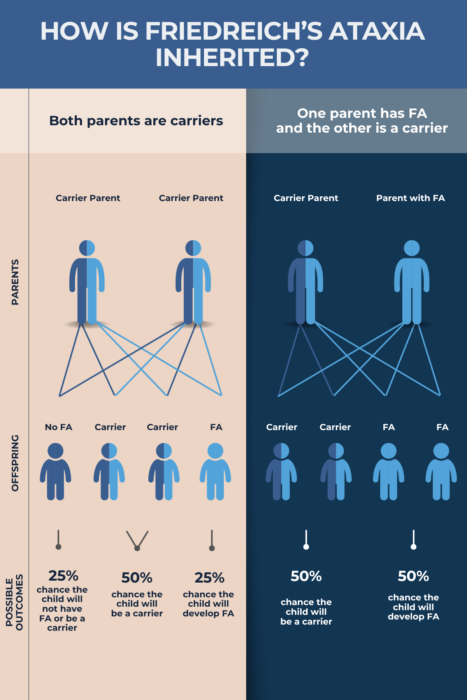
FA is inherited in what is called an autosomal recessive manner. Recessive means that the disease only will develop when a person inherits two mutated copies of the FXN gene, one from each biological parent. This may include having two copies of the gene with a GAA repeat expansion, or one copy with a repeat expansion and one with another type of mutation.
Inherited genetic information is contained in chromosomes, of which there are 23 pairs. Autosomal refers to the fact that the FXN gene is found on one of the 22 pairs of non-sex chromosomes (those other than X and Y), called autosomes. Specifically, it’s located on chromosome 9.
If a person inherits one normal copy of FXN and a mutated one, they are considered a disease carrier. This means they will not develop FA symptoms but will have a 50/50 chance of passing the mutation on to their biological children.
When two carriers have a biological child, there is a:
- 25%, or 1 in 4, chance that child will have FA
- 50%, or 1 in 2, chance the child will be a carrier
- 25%, or 1 in 4, chance the child will have two normal copies of FXN.
In turn, if a carrier conceives a child with someone with FA, there’s a 50% chance their offspring will develop FA and a 50% chance the child will be a carrier. Many people will not know they are disease carriers until they have a child with the disease.
Other risk factors
In the U.S., the risk of having FA is highest among people with Western European ancestry. FA is rarely identified in populations outside of Europe, the U.S., the Middle East, India, and North Africa.
Although the risk of FA inheritance is equal between men and women, research suggests that disease severity between men and women may differ. Particularly, some studies have indicated that women have differences in heart function that might protect against heart failure over time.
Friedreich’s Ataxia News is strictly a news and information website about the disease. It does not provide medical advice, diagnosis, or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.
Related articles
-
Discussion
-
 Discussion
Discussion
-
 Discussion
Discussion
-
 Discussion
Discussion
-
 Discussion
Discussion
-
Discussion