
Faulty Mitochondrial Process in Friedreich’s Ataxia Described
Written by |
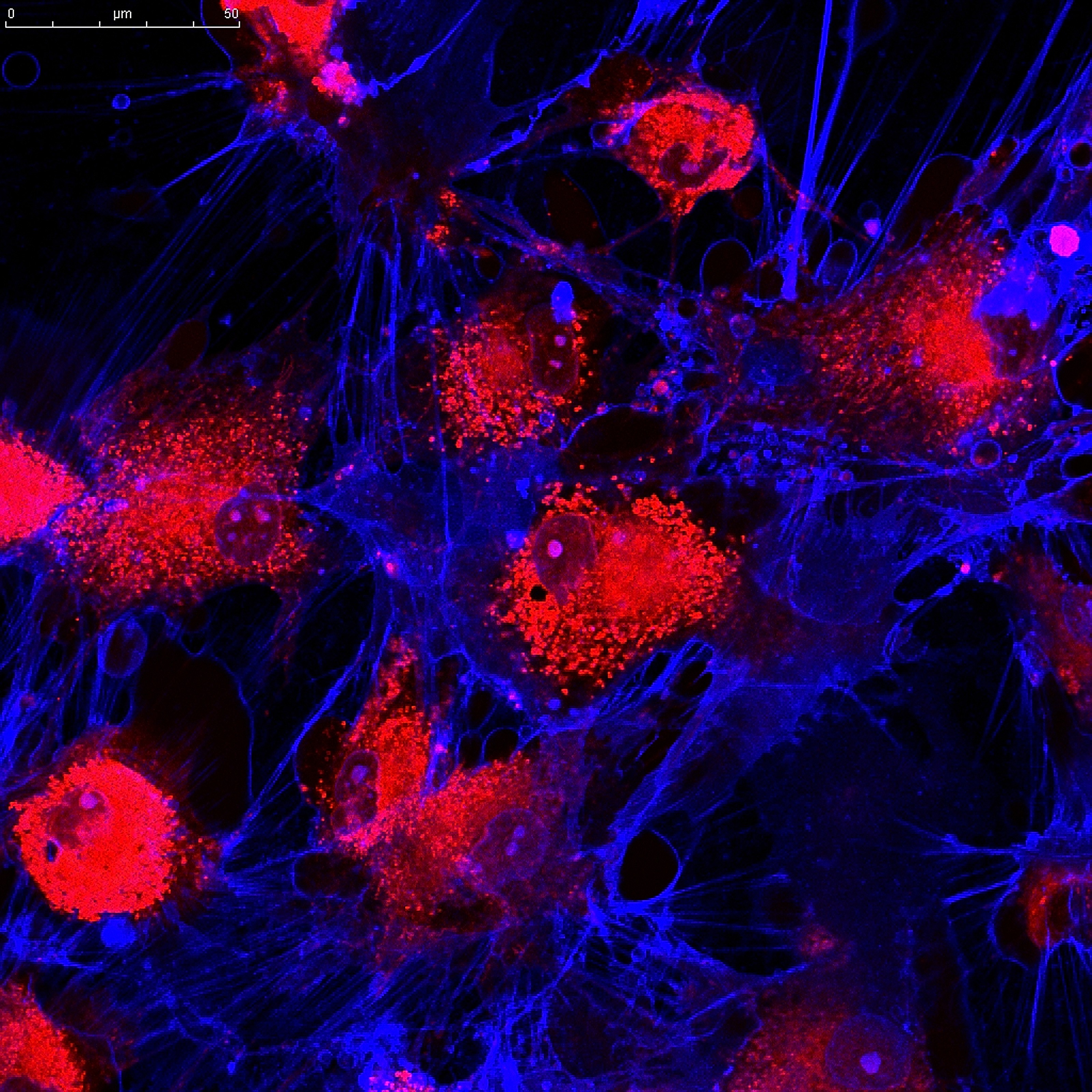



Scientists at University College London have, for the first time, described the full sequence of faulty mitochondrial processes in a Friedreich’s ataxia mouse model.
Findings show that dysfunctional mitochondria directly contribute to cell death through lipid peroxidation in disease-affected neurons – a degradation of cellular lipids that if prevented, stops brain cell death.
According to the National Institutes of Health, Friedreich’s ataxia is a rare inherited disease that causes nervous system damage and movement problems. It usually begins in childhood and leads to impaired muscle coordination (ataxia) that worsens over time.
Despite intense research into the mechanisms causing Friedreich’s ataxia, scientists are still in the dark about whether mitochondrial dysfunction is a side effect of accumulated iron, or if it is part of a primary process driving the disease.
Although earlier studies suggest that frataxin gene mutations lead to faulty mitochondrial respiration, which promotes the production of reactive oxygen species and brings about mitochondrial dysfunction, oxidative stress, and mitochondrial iron accumulation, technical issues limited the study’s reliability.
The team behind the new study, “Mitochondrial energy imbalance and lipid peroxidation cause cell death in Friedreich’s ataxia,“ argued that post-mortem tissue or fibroblast cells are poor models of the disease. Instead, they developed a cell culture model using neurons and glial cells from a validated mouse model of Friedreich’s ataxia that develops progressive disease with age.
Using live cell imaging and biochemical techniques, researchers found that despite similar low levels of frataxin in glial cells and a type of disease-affected neurons called cerebellar granule cells, the mitochondria were defective only in the neurons. In those cells, the lack of frataxin disrupted the mitochondrial membrane potential. The difference in electric charges between the in and outside of a cellular membrane is crucial for a mitochondrion’s ability to produce energy.
The publication in Cell Death and Disease, details how the disturbed potential emerged as a result of decreased complex I activity, accompanied by increased complex II activity. These complexes are part of the energy-producing mitochondrial respiratory chain and need to be in sync for the process to be effective.
When the balance between the two complexes became disturbed, researchers found that mitochondria produced more reactive oxygen species, which depleted the stores of the antioxidant glutathione and led to a 10-fold increase in lipid peroxidation in the cerebellar granule cells.
It became obvious that the lipid peroxidation was a key driver of cell death, since preventing lipids from becoming oxidized also prevented cell death. The strategy could be explored as a new treatment for Friedreich’s ataxia.



Leave a comment
Fill in the required fields to post. Your email address will not be published.