
Case study: Female patient with progressive imbalance, muscle weakness, and skeletal deformity
Written by Susan Perlman, MD
May 15, 2024
The patient is a 42-year-old, right-handed Caucasian female, followed for 20 years with slowly progressive imbalance, lower extremity weakness, scoliosis, foot deformities, and bladder dysfunction.
Physical exam, lab results, and medical history
The patient has had a long history of neurological problems beginning at the age of 12, when she was noted to have scoliosis.
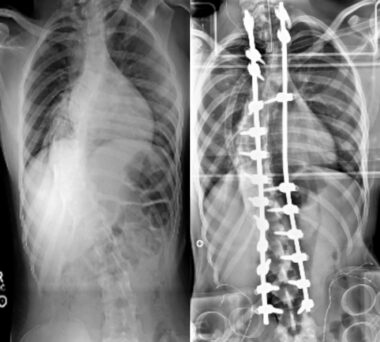
X-ray images showing patient’s scoliosis before (left) and after (right) corrective surgery. (Photos and images courtesy of Susan Perlman, MD).
Initially, the patient reported no difficulty with her walking, although the parents noticed the abnormal twisting of the hip when she walked during that time, felt to be due to the scoliosis. Since then, she has started to notice the problem with her walking, described as gait being unsteady, side to side, front and back, lacking rhythmicity.
The patient had episodes of falling, causing bone fractures. In addition, she described lacking sensation in her feet up to the mid-calf area bilaterally. She complained of impaired hand coordination with fine motor movement.
She also complained of slurred speech, which had become more evident during the years prior to her initial visit, but no swallowing issues. There was mild blurring of vision and fatigability. These issues had begun to affect her activities of daily living.
Her gait problem had been slowly progressive, and at initial visit at age 22, she was able to walk only with one person’s assistance and preferred to use a wheelchair. She had seen a physical therapist. She had undergone spinal fusion for the scoliosis at age 13. She was using ankle-foot orthoses for pes cavus. She was on no medications or vitamin supplements.
The patient has been seen by multiple neurologists, and excellent laboratory investigation has been performed. She had been told that she had deficits consistent with Charcot-Marie-Tooth disease and was referred to us to confirm this diagnosis.
Family history
She was born normally and she had had normal development. She did not have any neurological symptoms or developmental regression before the age of 12. There is no family history of any neurological conditions, and she has a brother who is two years younger and was well. One of her cousins on the paternal side was born with spina bifida.
Detailed medical history and examination
Her exam at her initial visit at age 22 showed she was alert and oriented to time, place, and person. Her Mini-Mental Status Examination was 30/30. She had a mild degree of bulbar dysarthria with mild slurring. There was no aphasia.
On cranial nerve examination, her pupils were 5 mm, both reactive to light. Direct fundoscopy did not reveal any evidence of optic atrophy. Eye movement examination revealed some fixation instability in primary gaze. She had sustained gaze-evoked nystagmus to the right. There was no torsional component noted. She had no complaints of vertigo.
Facial sensation was symmetrical bilaterally. She had a mild degree of bilateral facial weakness, more evident in the orbicularis oris around her mouth area. Her tongue was in the midline, and she had a bilateral palatal elevation symmetrically. The uvula was midline.
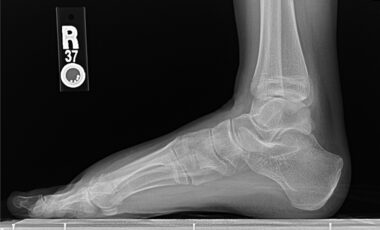
X-ray image showing pes cavus deformity in patient’s foot
Her motor examination revealed normal tone in all four extremities. There was a mild degree of muscle wasting in the small muscles of the hands and the intrinsic foot muscles. She had a pes cavus deformity in bilateral foot area, but there were no hammertoes. There was some degree of wasting of the gastrocnemius, although it was mild. There was no evidence of the inverted champagne bottle appearance in her calf area. There was a mild degree of weakness distally of 4+/5.

Patient typing showing mild hand deformity
She clearly had a finger-to-nose dysmetria and heel-to-shin dysmetria. There was no titubation. She had a mild degree of kinetic tremor when she approached the target on the finger-to-nose test.
Sensory examination revealed stocking sensory loss for pinprick and light touch in the lower extremities, up to about the ankle area. She had a clear impairment of proprioception and vibration sense in the lower extremities.
Reflex examination revealed diffuse areflexia, with upgoing plantar response on the left and mute plantar response on the right.
On gait examination, she walked with a wide-based gait and required one assistant on her side. She would fall if she had nobody to hold onto or rails to hold onto during her walking. She was not able to perform tandem walk. She was not able to perform Romberg test due to her gait unsteadiness.
Prior testing included:
- MRI of the brain at age 22 was reported as normal, although cervical cord atrophy was seen on MRI of the spinal cord.
- She had a nerve conduction study and EMG performed shortly after her foot deformity and lower extremity sensorimotor changes were seen. This led to a muscle biopsy of the left gastrocnemius at age 14, which was reported to show type 1 predominant chronic denervation. A sural nerve biopsy around the same time showed large myelinated fiber loss.
- Blood work was normal and included basic hematologic and biochemical measures; vitamins B1, B12, E, and folic acid serum concentration testing; sedimentation rate, CK levels, urine amino acids concentration, urine organic acids profile, serum lactic and pyruvic acids concentration, very-long-chain fatty acids concentration, alpha-fetoprotein, thyroid hormone assay, and urine heavy metals.
- No genetic testing had been performed.
The phenotypic presentation of this patient included childhood onset caudal to rostral spinocerebellar ataxia progression, posterior column sensory loss, areflexia with bilateral upgoing toes, scoliosis, and pes cavus.
Based on her phenotypic presentation, this was consistent with Friedreich’s ataxia, the most common autosomal recessive hereditary neurological ataxia presenting between the ages of 5 and 15 (2 in 100,000), leading to wheelchair dependence in most cases by age 20. There would not necessarily be a family history of ataxia in this recessive disorder nor any risks associated with the carrier state.
Charcot-Marie-Tooth is a common condition that often mimics Friedreich’s ataxia, especially if there are early neuromuscular or orthopedic changes.
Inborn errors of metabolism that could cause ataxic or neuromuscular features had been ruled out in this patient. Brain imaging ruled out a primary cerebellar disorder (genetic or acquired), white matter disease, or space-occupying lesion.
We requested genetic tests for the GAA expansion seen in the first intron of the frataxin protein gene. She tested positive and was homozygous for two alleles with approximately 600 GAA repeats each, confirming a genetic diagnosis of Friedreich’s ataxia.
Diagnostic review
The GAA expansion reduces the genetic transcription and translation of frataxin protein, which is important for normal mitochondrial functioning. The size of the mutation on the smaller of the two alleles determines how much frataxin protein might be produced — a smaller mutation allowing more protein production with later onset, slower progression, and fewer extraneural features; larger mutations (>500 GAA repeats) being associated with a greater risk of extraneural features (orthopedic complications — scoliosis, pes cavus, hypertrophic cardiomyopathy, diabetes, and vision and hearing loss). Compound heterozygotes (one GAA expansion and one point mutation) could present with a more severe and earlier onset phenotype or a milder and later onset phenotype.
Treatment discussion and outcome
For the newly diagnosed patient with Friedreich’s ataxia, we recommend these health maintenance procedures:
- Annual visit to a neurologist to monitor ataxia and neuromuscular features
- Annual visit to a cardiologist for a heart check (ECG, echocardiogram)
- Annual visit to an orthopedist for a spine and foot check
- Annual blood test to screen for diabetes (fasting blood sugar or hemoglobin A1c; urine sugar test)
- Baseline vision and hearing test
- Evaluation with physical therapy and occupational therapy about home exercise
- Any use of antioxidant supplement (vitamin E 400 IU each day, coenzyme Q10 400 mg each day, or Idebenone 500 mg capsules three times per day with meals) should be discussed with doctor prior to starting.
- Consider participation in research opportunities (natural history studies, biomarker studies, drug trials).
- Have patients sign up for the Friedreich’s Ataxia Global Patient Registry to receive notices about new research studies in which they can participate.
- Genetic counseling to discuss risks for other family members and reproductive options.
- Good resources for more information: Friedreich’s Ataxia Research Alliance; Friedreich’s ataxia Parent’s Group; and the National Ataxia Foundation.
Medications can be used to treat bothersome symptoms (muscle cramps and spasms, orthopedic pain, sleep disorders, bladder and bowel dysfunction, and depression or anxiety).
Fatigue may be an ongoing issue, caused by the ataxia itself, neuromuscular weakness, deconditioning, sleep disorders, or depression. Treatment of these underlying causes can be helpful. Amantadine has been used off-label for ataxia, tremor, and fatigue, but may be constipating.
With the newly FDA-approved drug Skyclarys (omaveloxolone), all patients with Friedreich’s ataxia aged 16 years and older should be encouraged to try it for at least 6-12 months. It can be symptomatically helpful for ataxia and may slow progression of symptoms for at least two to three years. It requires monitoring of liver function, lipid profile, and BNP monthly for at least the first three months.
Side effects (headache, muscle pain, abdominal pain, nausea, and fatigue) usually resolve in the first one to two months of use, but occasionally might require a reduction in dosage from 150 mg per day to 100 mg per day. Although the FDA placed the lower age of 16 as the only restriction, access to the drug might be challenged by payors on the basis of the other inclusion/exclusion criteria of the MOXIE study (ambulatory ability, cardiac status, presence of pes cavus). Patient assistance programs are available.
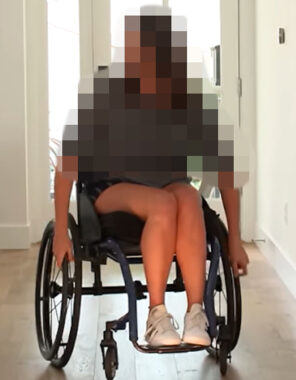
The patient has progressed to complete wheelchair dependence.
The patient profiled above has progressed to complete wheelchair dependence, but has not developed cardiomyopathy or diabetes (the combination being responsible for premature death before the age of 40 in many patients).
She is independent for transfers. Her current medications are Baclofen, 10 mg three times daily, for muscle spasms; Pantoprazole, 40 mg daily; Solifenacin, 10 mg daily for neurogenic bladder, and probiotics for bowel function.
Over the past year, she has noticed some worsening of vision and hearing. Speech is more effortful and phlegm tends to build up in her throat. She may cough with liquids. Writing and fine hand manipulation are more difficult.
She has been on Skyclarys for three months with stable laboratory values and mild stomach upset that has resolved. Her family feels that her speech is improved on Skyclarys and the patient feels she has better trunk control and it is easier to transfer.
Video commentary
Quiz

Sources
Omaveloxolone for the treatment of Friedreich ataxia: clinical trial results and practical considerations. Lynch DR, Perlman S, Schadt K. Expert Rev Neurother. 2024 Mar;24(3):251-258.
Friedreich ataxia: clinical features and new developments. Keita M, McIntyre K, Rodden LN, Schadt K, Lynch DR. Neurodegener Dis Manag. 2022 Oct;12(5):267-283.
Friedreich Ataxia: Multidisciplinary Clinical Care. Lynch DR, Schadt K, Kichula E, McCormack S, Lin KY.J Multidiscip Healthc. 2021 Jun 28;14:1645-1658.
Three Adult-Onset Autosomal Recessive Ataxias: What Adult Neurologists Need to Know. Paulus-Andres JA, Burnett MS. Neurol Clin Pract. 2021 Jun;11(3):256-262.
About Susan Perlman, MD
Susan Perlman, MD, is a clinical professor of neurology and the director of the Ataxia Center at the UCLA Medical Center in Los Angeles. Her specialty is ataxias, including Friedreich’s ataxia. She is also the director of clinical trials in UCLA’s Program in Neurogenetics and sits on the Medical and Research Advisory Board at the National Ataxia Foundation.
 David Lynch, MD, PhD, is the director of the Friedreich’s Ataxia Program at Children’s Hospital of Philadelphia.
David Lynch, MD, PhD, is the director of the Friedreich’s Ataxia Program at Children’s Hospital of Philadelphia.